A review study: Computational techniques for expecting the impact of non-synonymous single nucleotide variants in human diseases
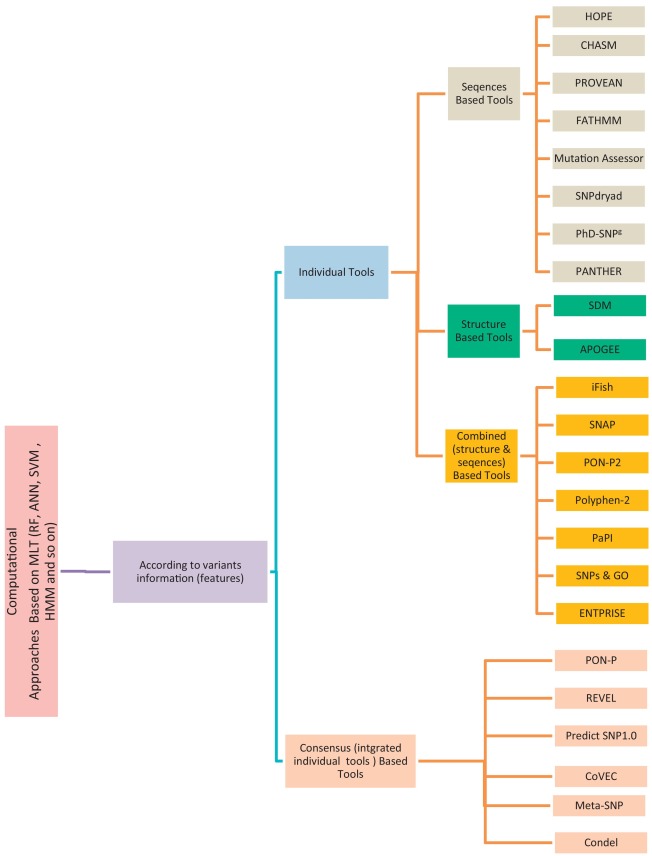
Non-Synonymous Single-Nucleotide Variants (nsSNVs) and mutations can create a diversity effect on proteins as changing genotype and phenotype, which interrupts its stability. The alterations in the protein stability may cause diseases like cancer. Discovering of nsSNVs and mutations can be a useful tool for diagnosing the disease at a beginning stage. Many studies introduced the various predicting singular and consensus tools that based on different Machine Learning Techniques (MLTs) using diverse datasets. Therefore, we introduce the current comprehensive review of the most popular and recent unique tools that predict pathogenic variations and Meta-tool that merge some of them for enhancing their predictive power. Also, we scanned the several types computational techniques in the state-of-the-art and methods for predicting the effect both of coding and noncoding variants. We then displayed, the protein stability predictors. We offer the details of the most common benchmark database for variations including the main predictive features used by the different methods. Finally, we address the most common fundamental criteria for performance assessment of predictive tools. This review is targeted at bioinformaticians attentive in the characterization of regulatory variants, geneticists, molecular biologists attentive in understanding more about the nature and effective role of such variants from a functional point of views, and clinicians who may hope to learn about variants in human associated with a specific disease and find out what to do next to uncover how they impact on the underlying mechanisms. © 2018